Defectos Congénitos de la Glicosilación de Proteínas
El Comité de Redacción de Acta Bioquímica Clínica Latinoamericana ha seleccionado este artículo publicado en INFORME ALAC, Año XII - Nº 1 – 2007, para su difusión a través de FABA Informa:
Los defectos congénitos de la glicosilación (Congenital Disorders of Glycosylation CDG), constituyen un grupo de enfermedades genéticas de transmisión autosómica recesiva.
Bioquímicamente, se caracterizan por alteraciones que se producen en el enlace, o en el procesamiento de los oligosacáridos, los cuales tienen funciones muy importantes en la función, metabolismo y estructura de las glicoproteínas y otros glicoconjugados.
La mayor parte de las proteínas extracelulares, las proteínas de membrana y varias intracelulares están glicosiladas, por lo tanto un defecto en este proceso confiere a este grupo de patologías una gran importancia.
Niels A. F. Suldrup (A)
IACA Laboratorio Metabolopatías – San Martín 68 (8000)
Bahía Blanca– Buenos Aires - Argentina
Los defectos congénitos de la glicosilación o CDG, se describieron por primera vez en 1980 y durante años se los denominaron “síndromes de glicoproteínas deficientes en carbohidratos”. En los 25 años siguientes se han descripto más de 30 patologías asociadas o producidas por los CDG, clasificadas y subclasificadas en varios tipos de acuerdo con la falla enzimática presente.
Junto a las patologías mitocondriales, los CDG son las enfermedades metabólicas más complejas. Ambas son enfermedades multisistémicas, de presentación clínica variable y generalmente involucran tanto al sistema nervioso central como al periférico.
Fundamentos de la glicosilación enzimática de proteínas
La glicosilación de proteínas es la forma más compleja de modificación de biomoléculas que existe. Consta básicamente de dos etapas, la primera es la síntesis de los oligosacáridos y la segunda es la unión de estas moléculas a una proteína (glicoproteína) o a lípidos (glicolípidos, esfingolípidos).
Las glicoproteínas se clasifican de acuerdo con el tipo de unión covalente que existe entre el polipéptido y el oligosacárido. Se conocen tres tipos de glicoproteínas:
a) N-Glicoproteínas: La unión se lleva a cabo sobre el nitrógeno del grupo amido del aminoácido asparagina.
b) O-Glicoproteínas: La unión se realiza sobre el oxígeno del grupo hidroxilo de los aminoácidos serina o treonina,
c) C-Glicoproteínas: La unión es a través del carbono 2 del grupo indol del aminoácido triptofano.
La síntesis de los N-glicanos es una vía metabólica compleja que incluye unos 40 pasos enzimáticos, que se producen en tres compartimientos celulares: en el citoplasma, en el retículo endoplásmico y en el aparato de Golgi. Este proceso se puede esquematizar en tres fases concatenadas (Figura 1).
En el citosol se forma el compuesto nucleótido-azúcar, generalmente, guanosina difosfato-manosa (GDP-Man), pero también puede ser UDP-glucosa y UDP-N-acetilglucosamina (pasos 1 a 4 de la Figura 1 ). Este compuesto se une al dolicolfosfato y de esta manera ingresa al retículo endoplásmico (RE) (pasos 5 a 8), donde por medio de glucosiltransferasas específicas se agregan secuencialmente distintos glúcidos, hasta formar un oligosacárido común de 14 residuos monosacáridos.
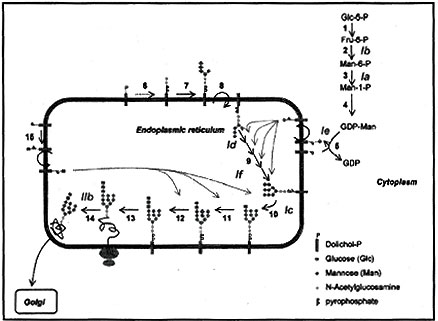
< Figura 1:Biosisntesis de los glicanos y glicosilacion enzimatica. (Extraido de Pediatric Research vol 52 Nº 5 2002)
Esta molécula es tomada, entonces, por el complejo enzimático oligosacaridiltransferasa (paso 13), que reconoce una secuencia consenso formada por los aminoácidos Asparagina-X-Serina o Asparagina-X-Treonina, donde X es cualquier aminoácido (excepto prolina) y lo transfiere al N amido de la asparagina.
Esta proteína glicosilada es procesada en el RE y en el aparato de Golgi sufre modificaciones mediante la eliminación de residuos glucosa y manosa, y la adición de otras moléculas, como ácido siálico, N-acetilglucosamina, fucosa o galactosa.
La biosíntesis de los O-Glicanos se produce mayormente en el aparato de Golgi sobre la proteína ya sintetizada.
No existe una secuencia consenso de aminoácidos y forman los denominados O-xilosilglicanos, O-manosilglicanos y O-N-acetilgalactosaminilglicanos.
Si bien este parece un mecanismo más sencillo, el número de O-glicanos es muy superior al de N-glicanos.
Ambos procesos de glicosilación enzimática otorgan a las proteínas estabilidad y función. Se producen cientos de glicoproteínas distintas con un alto grado de complejidad y especificidad que les permiten modular y regular una infinidad de procesos celulares, tales como resistencia a proteasas, estructura, desarrollo, migración, diferenciación, adhesividad, reconocimiento celular y antigenicidad.
Defectos en la N-glicosilación de proteínas
Desde este primer caso reportado en los años 80 por Jaecken y col. mediante la observación de dos gemelas con retraso mental y el estudio de determinadas proteínas séricas, hasta la fecha se han descripto más de 20 enfermedades.
Para su estudio, los CDG se dividen en dos grupos basados en las características bioquímicas y según la principal etapa deficiente.
En los defectos de glicosilación de tipo I (CDG-I) la falla metabólica ocurre en el citoplasma o en el retículo endoplásmico, ruta de elongación, e incluye a las enfermedades de enlace de los N-glicanos.
Los defectos de glicosilación de tipo II (CDG-II) ocurren en la etapa de procesamiento de los N-glicanos, en el retículo endoplásmico y en el aparato de Golgi.
En la Tabla I se muestra esta clasificación con su respectivo defecto enzimático y los sistemas u órganos mayormente afectados.
(Ver Tabla I y II )
Diagnóstico de los defectos en la N-glicosilación
Debido a que los CDG afectan a varias o a todas las estructuras del organismo, los síntomas y la severidad varían con cada tipo de defecto, además de tener variaciones propias individuales.
La mayoría de los tipos de CDG están asociados con dimorfismos faciales y corporales menores, trastornos neurológicos, retraso en el crecimiento.
Algunos pueden presentar coagulopatías, hepatopatías y problemas intestinales.
El diagnóstico de estas patologías se realiza ante la sospecha clínica y se confirma por la evidencia bioquímica de anomalías en la glicosilación de ciertas proteínas.
Para realizar el diagnóstico diferencial de los CDG, la glicoproteína que se utiliza como marcadora es la transferrina.
La transferrina (TRF) está presente en el suero en grandes concentraciones y cada
molécula de TRF tiene unidas dos cadenas que presentan dos, tres y cuatro ramificaciones de
glicanos, las cuales a su vez presentan 2, 3, 4, 5 y 6 residuos de ácido siálico (Figura 2).
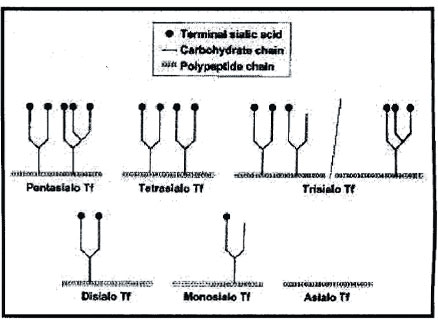
< Figura 3 Estructura de las isoformas de Transferrina serica mas comunes (extraido de Clinical Chemistry vol 47 Nº 7, 2001)
La forma mayoritaria de las transferrinas séricas es la tetrasialo, es decir, la TRF que tiene dos cadenas con dos ramificaciones (biantenaria) y cada una con 2 residuos sialo. Las isoformas menos sialiladas, asialo (ningún residuo sialo), monosialo, disialo y trisialo-TRF son minoritarias y en conjunto se las denomina Transferrina Carbohidrato Deficiente (CDT).
Cuando se produce un defecto en la glicosilación de la TRF, ésta incorpora menos ácido siálico a su molécula, lo que provoca un incremento en la concentración sérica de la CDT.
La determinación de CDT, ya sea en valor absoluto o relativo a la TRF total, puede utilizarse como método de screening para los CDG en niños con sospecha de enfermedad metabólica.
El valor de CDT % para la población pediátrica según distintas revisiones, se ha establecido entre 2,5 y 3 % sin distinción de sexo ni edad. Por lo tanto, un valor superior al 3 % indicaría un paciente con probable CDG.
El estudio del patrón de las isoformas de transferrina (sialotransferrinas) producido por una corrida electroforética con enfoque isoeléctrico (IEF), o por medio del patrón de elusión de una corrida de electroforesis capilar (Figura 3), son definitivamente informativos.
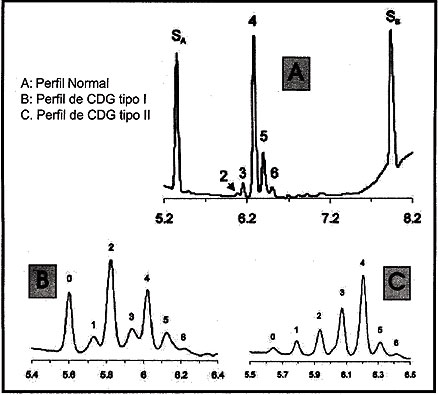
< Figura 3: Perfil2es de electroforesis capilar de transferrina sérica
La deficiencia parcial de ácido siálico en el azúcar terminal provoca un cambio catódico. Se reconocen dos tipos principales de patrones de este cambio.
El tipo 1 es caracterizado por un aumento en las isoformas asialo y disialo transferrinas y una disminución de las formas tetra, penta y hexa sialo, lo que indica una pérdida de glicanos completa, típica de los pacientes con CDG de tipo I.
El otro patrón de corrida presenta un incremento en las formas monosialo y trisialo lo que indica la existencia de glicanos estructuralmente anómalos, demostrando una falla en el procesamiento de los glicanos y orientando el diagnóstico hacia un CDG de tipo II.
El análisis porcentual de cada una de estas fracciones también brinda una valiosa información para la orientación sobre el subtipo de cada enfermedad.
La confirmación definitiva se realiza mediante la determinación de la actividad de cada enzima específica. Los análisis moleculares no son muy útiles debido a la gran cantidad de mutaciones que se han encontrado (p.ej., para la PMN2 se conocen más de 35 mutaciones).
De todos los defectos de glicosilación, el tipo la (CDG-la o deficiencia de fosfomanomutasa 2, PMN2) es el más comúnmente observado, con más del 70 % de todos los casos reportados de CDG.
La sintomatología puede ser reconocida a los pocos días del nacimiento, el sistema nervioso está afectado en todos los pacientes, y se observa estrabismo, movimiento anormal de los ojos, hipotonía, ataxia e hiporreflexia.
En la infancia presentan retraso psicomotor, distribución anormal del tejido adiposo subcutáneo (fat pads), episodios de shock, retinitis pigmentosa y epilepsia.
El defecto de glicosilación de tipo lb o deficiencia de fosfomanosa isomerasa, se detectó por primera vez en 1998.
Esta enfermedad no se presenta con deterioro del sistema nervioso, solamente afecta al hígado e intestinos y es la única que tiene un tratamiento eficaz. La terapia consiste en la administración de manosa por vía oral varias veces por día.
Los síntomas clínicos revierten y los parámetros de laboratorio de evaluación hepática se normalizan rápidamente, pero la corrección del patrón de transferrinas puede tardar varios meses.
Defectos de la O-glicosilación de proteínas
Los desórdenes congénitos que afectan a la O-glicosilación forman un grupo de enfermedades que no tienen un nombre colectivo, por lo tanto no es fácil realizar una revisión rápida de ellas.
Cada vez se está haciendo más evidente que el defecto no necesariamente se halla en una enzima específica, sino que puede encontrarse en la biosíntesis del nucleótido-azúcar, en su transporte al retículo endoplásmico, en el pasaje del RE al aparato de Golgi o en el procesamiento que se produce en el aparato de Golgi.
La función de las O-glicanos está muy relacionada con la estructura y la estabilidad de la proteína, aspectos inmunes, modulación enzimática y funcionalidad de la proteína.
Por ejemplo, los O-glicanos tipo mucina son muy importantes. Las mucinas son proteínas altamente O-glicosiladas y están presentes en todas las superficies húmedas del cuerpo, como el tracto digestivo, respiratorio y génito-urinario. Estas mucinas tienen la capacidad de fijar el agua y producir geles (mucus) que además tienen propiedades antibacterianas.
Otra función, es el reconocimiento entre proteínas e interacción con las lectinas. Muchos procesos celulares están influenciados por los azúcares que captan las lectinas, tales como, crecimiento celular, apoptosis y distintas interacciones célula-célula, fertilización del oocito, antígenos de los grupos sanguíneos ABO y el reconocimiento de glicopéptidos por el complejo mayor de histocompatibilidad.
Errores congénitos que afectan la O-glicosilación de proteínas
Las enfermedades que afectan a la biosíntesis de proteínas O-glicosiladas se describieron por primera vez en 1990 y desde entonces constituyen un grupo de enfermedades en expansión.
El primer defecto descripto fue el de la enzima ?-1,4-galactosiltransferasa 7 que afecta a la síntesis de los glucosaminoglicanos (O-xilosilglicanos) y que es la causante de la variante progeroide del síndrome de Ehlers-Danlos.
En esta misma vía metabólica también se encuentra la enzima heparánsulfato copolimerasa cuya deficiencia causa el síndrome de exostosis múltiple hereditario.
Este último es el único CDG conocido que tiene herencia autosómica dominante.
Durante los últimos años se han identificado una serie de desórdenes en las rutas metabólicas de los O-manosilglicanos que serían responsables de algunos tipos de distrofias musculares congénitas.
Por último, los defectos de la O-glicosilación de los glicanos tipo mucina, causados por el déficit de N-acetilgalactosaminil transferasa 2 provoca la enfermedad llamada Calcinosis Tumoral Familiar, en la que se producen depósitos de calcio en la piel y tejido subcutáneo.
En la Tabla II se pueden ver varias enfermedades genéticas humanas debidas a defectos en la O-glicosilación de proteínas.
Defectos en la C-glicosilación de proteínas
La C-manosilglicosilación difiere de los anteriores procesos fundamentalmente debido a que involucra una unión covalente entre un residuo manosilpiranosil y el C2 del grupo indol del triptofano mediante un enlace carbono-carbono. Este tipo de unión fue descripto por primera vez por Hofsteenge en 1994 en la enzima RNasa 2, presente en la orina humana.
Esta enzima es una proteína de secreción y contiene una secuencia señal para atravesar las membranas del Retículo Endoplásmico.
Estudios posteriores demostraron que la C-glicosilación es un mecanismo presente en varios organismos, tales como hongos, aves y mamíferos.
Si bien aún no se han identificado patologías directamente relacionadas con defectos en la biosíntesis de C-glicoproteínas, estas estructuras de glicanos unidos al triptofano se han encontrado en diversas proteínas humanas, algunas relacionadas con la angiogénesis, con la migración neuronal, con el sistema inmune, metaloproteasas y receptores de membrana.
El descubrimiento de la C-glicosilación es una importante contribución al estudio de las glicoproteínas. Las modificaciones post-traducción que sufren las proteínas son esenciales para su estructura y función.
El estudio del la C-manosilación es un complemento útil en el campo de la producción de proteínas recombinantes.
Correspondencia y Consultas:
Niels A. F. Suldrup:
metabolopatias@iaca.com.ar
IACA Laboratorios - Metabolopatías
San Martín 68 (B8000FIB) Bahía Blanca, Buenos Aires. Argentina
Bibliografía
• Jaeken J, Matthijs G, Carchon H, Van Schaftingen E. Defects of N-glycan synthesis. In: Scriver CR, Beaudet AL Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease, 8th ed. New York: McGraw-Hill:1.601-1.622 (2001) .
• Jaeken J. Congenital disorders of glycosylation In: Fernandes J, Saudubray JM, van den Berghe G, Walter J editors Inborn Metabolic diseases, 4 th ed. Germany: Springer: 525-530 (2006).
• Butler M, Quelhas D, Critchley AJ, Carchon H, Hebestreit HF, Hibbert RG, Vilarinho L, Teles E, Matthijs G, Schollen E, Argibay P, Harvey DJ, Dwek RA, Jaeken J. Detailed glycan analysis of serum glycoproteins of patients with congenital disorders of glycosylation indicates the specific defective glycan processing step and provides an insight into pathogenesis. Glycobiology 13: 601-622 (2003).
• Grünewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation: A review Pediatr Res 52: 618-624 (2002).
• Helander A, Bergstrom J, Freeze H. Testing for Congenital Disorders of Glycosylation by HPLC Measurement of Serum Transferrin Glycoforms. Clin Chem 50: 954-958 (2004).
• Wuyts B, Delanghe JR, Kasvosve I, Wauters A, Neels H. Janssens J. Determination of carbohydrate-deficient transferring using capillary zone electrophoresis. Clin Chem 47: 247-255 (2001).
• Carchon H, Chevigné R, Falmagne JB, Jaeken J. Diagnosis of congenital disorders of glycosylation by capillary zone electrophoresis of serum transferrin. Clin Chem 50: 101-111 (2004).
• Charlwood J, Clayton P, Keir G, Mian N, Winchester B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology 8: 351-357 (1998).
• Whitfield JB. Transferrin isoform analysis for the diagnosis and management of hazardous or dependant drinking. Clin Chem 48: 2.095-2.096 (2002).
• Wopereis S, Lefeber D, Morava E, Wevers R. Mechanism in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: A review. Clin Chem 52: 574-600 (2006).
• Jaeken J, Matthijs G, Saudubray J, Dionisi-Vici C, Bertinl E, DeLonlay P, et al. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-Intestinal presentation. Am J Hum Genet 62: 1.535-1.539 (1998).
• Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet 360: 1.419-21 (2002).
• Wopereis S, Morava E, Grünewald S, Adamowicz M, Huijben K, Lefeber D, Wevers R. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycoblology 15: 1.312-1.319 (2005).
|
|