¿En
vísperas de tratamientos eficaces para la enfermedad de Alzheimer?
En el curso de 2006 se conocieron
los resultados de una serie de investigaciones con ratones modificados
genéticamente para expresar las lesiones cerebrales de la
enfermedad de Alzheimer. Estos estudios abren caminos para el futuro
desarrollo de tratamientos eficaces para esta enfermedad, que hasta
ahora es incurable.
El Comité de Redacción
de Acta Bioquímica Clínica Latinoamericana ha seleccionado
este artículo publicado en la CIENCIA HOY
Volumen 17 Nº 97 – 2007, para su difusión a través
de FABA Informa.
<
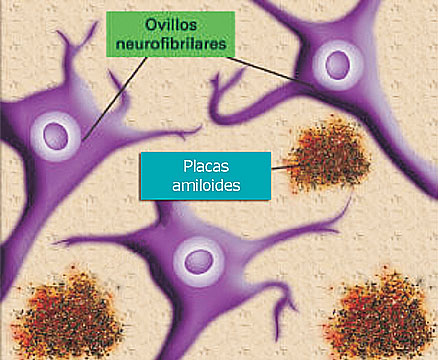
Fig. 1.- Lesiones características
del cerebro en la enfermedad de Alzheimer
La enfermedad de Alzheimer es la causa más frecuente de la
demencia senil. Entre el 2 y el 5% de las personas de 65 años,
y el 25% de quienes superan los 85 años de edad la padece.
El progresivo aumento de la expectativa de vida de la población
determina que el Alzheimer tenga una creciente incidencia en los
gastos en salud. En la actualidad la atención de la enfermedad
consume anualmente unos 145 mil millones de dólares.
Síntomas y signos
La enfermedad comienza con síntomas casi imperceptibles.
Un ejemplo de esto es el caso de la novelista británica Iris
Murdoch (1919-1999). Un estudio publicado en 2005 en la revista
Brain demostró que en la última novela de Murdoch
(Jackson’s Dilemma) ya era detectable el déficit cognitivo
propio de la enfermedad de Alzheimer que le fuera diagnosticada
a su autora luego de que terminara de escribirla.
La enfermedad se caracteriza por la pérdida de la memoria
(Ronald Reagan, otra de sus víctimas, no recordaba que había
sido presidente de los Estados Unidos), de la iniciativa y del pensamiento
abstracto. A medida que avanza, desaparece la capacidad de hablar
y de realizar movimientos coordinados.
Los pacientes manifiestan una desorientación que puede conducir
a que se extravíen aun en lugares que le han sido familiares
por años. De hecho, las averiguaciones de paradero de ancianos
extraviados que difunden los medios suelen corresponder a pacientes
con Alzheimer. Durante el curso de la enfermedad suelen aparecer
agitación, insomnio y depresión, los que en general
no responden a los psicofármacos que son eficaces en enfermedades
diferentes al Alzheimer. A medida que la enfermedad se acerca a
su etapa final quienes la padecen pierden toda capacidad de valerse
por sí mismos. La dolencia no sólo afecta al paciente
sino que también influye sobre sus seres queridos, que se
ven obligados a contemplar impotentes el lento pero inexorable derrumbe
de la personalidad del enfermo.
Lesiones cerebrales en la enfermedad
de Alzheimer
El cerebro de los pacientes con Alzheimer presenta una serie de
alteraciones que conducen a la destrucción de neuronas y
a la pérdida de las conexiones que las vinculan. Estas lesiones
incluyen:
la formación de agregados extracelulares llamados placas
amiloides, el depósito en el interior de las neuronas de
ovillos neurofibrilares y, la acumulación en las neuronas
de proteínas unidas a la ubiquitina. La ubiquitina es una
pequeña proteína que está presente en todas
las células (de ahí su nombre) que se une a otras
permitiendo así que éstas sean reconocidas por los
sistemas encargados de su degradación.
Las placas amiloides se forman debido a precipitación de
agregados de un péptido de entre 39 y 43 aminoácidos
conocido como amiloide beta (Aâ)). Este péptido es
un pequeño fragmento de una proteína llamada APP (de
Amyloid Precursor Protein) formada por una única cadena de
753 aminoácidos que atraviesa de lado a lado la membrana
celular. El péptido Aâ se desprende de la APP por la
acción sucesiva de las enzimas beta y gama secretasas
(Figura 2). Este péptido es un
componente normal del organismo, pero en condiciones patológicas,
como en la enfermedad de Alzheimer, adquiere una configuración
alargada la que permite a sus moléculas asociarse entre sí
formando agregados insolubles que conducen a las placas amiloides.
Este proceso requiere otras sustancias, algunas de ellas se mencionarán
más abajo al describir posibles tratamientos de la enfermedad.
Muchos estudiosos creen que el péptido Aâ o alguno
de sus derivados -y no la placa amiloide en sí- es el principal
causante de las alteraciones cerebrales propias de la enfermedad
de Alzheimer. También se sabe que de los distintos péptidos
Aâ, el que contiene 42 aminoácidos es el principal
responsable de las lesiones. A pesar de que se origina fuera de
las células, algunos de los efectos neurotóxicos pueden
requerir la penetración del péptido Aâ en las
neuronas (figura 2).
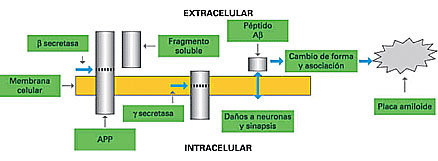 <
Figura 2.
La proteína precursora del amiloide (AAP) atraviesa la membrana
que rodea las células. La â secretasa cataliza la escisión
de un fragmento soluble situado en la superficie extracelular de
la membrana; este fragmento no participa directamente en la génesis
de las lesiones de la enfermedad de Alzheimer. El corte que produce
la â secretasa deja en la membrana un fragmento que al ser
escindido por la ã secretasa da lugar al péptido Aâ.
En la enfermedad de Alzheimer este péptido se acumula, cambia
de forma y se asocia dando lugar a agregados insolubles que son
el principal componente de las placas amiloides. El diagrama no
muestra el camino llamado no amiloidogénico en el cual la
AAP es cortada primero por la ã secretasa dando un fragmento
que no produce péptidos Aâ. <
Figura 2.
La proteína precursora del amiloide (AAP) atraviesa la membrana
que rodea las células. La â secretasa cataliza la escisión
de un fragmento soluble situado en la superficie extracelular de
la membrana; este fragmento no participa directamente en la génesis
de las lesiones de la enfermedad de Alzheimer. El corte que produce
la â secretasa deja en la membrana un fragmento que al ser
escindido por la ã secretasa da lugar al péptido Aâ.
En la enfermedad de Alzheimer este péptido se acumula, cambia
de forma y se asocia dando lugar a agregados insolubles que son
el principal componente de las placas amiloides. El diagrama no
muestra el camino llamado no amiloidogénico en el cual la
AAP es cortada primero por la ã secretasa dando un fragmento
que no produce péptidos Aâ.
Los ovillos neurofibrilares (figura 1) son depósitos
intracelulares en los que participan agregados insolubles de proteínas
tau. Estas proteínas forman parte del citoesqueleto, esto
es, de la red de túbulos y fibrillas que da forma a las células
y que participa en su motilidad y su división.
La acumulación de proteínas unidas a la ubiquitina
se debe a que en el Alzheimer la actividad de una enzima propia
de las neuronas llamada Uch-L 1 (hidrolasa C Terminal de la ubiquitina)
está disminuida, probablemente por acción del péptido
Aâ. Esta enzima es necesaria para la liberación de
la ubiquitina una vez que ha cumplido su función en las neuronas.
En búsqueda de nuevos tratamientos
El conocimiento de las lesiones cerebrales del Alzheimer ha servido
de base para la búsqueda de tratamientos eficaces de la enfermedad.
Esto ha sido facilitado por la creación de ratones que contienen
genes humanos que expresan las proteínas que se alteran en
la enfermedad. Estos ratones transgénicos desarrollan lesiones
cerebrales y trastornos de la memoria y el aprendizaje parecidos
a los de la enfermedad humana. Su utilización para la búsqueda
de tratamientos ha logrado resultados promisorios al aplicar los
siguientes tres enfoques:
• el bloqueo de los efectos del
péptido Aâ,
• la mejoría de la función
sináptica y,
• el restablecimiento de los niveles
normales de la Uch-L 1.
Bloqueo
de los efectos del péptido Aâ
Si los daños cerebrales del Alzheimer fueran causados por
la acumulación del péptido Aâ debería
ser posible evitarlos bloqueando los efectos de este péptido.
Para lograr esto, en una etapa inicial se ensayaron las llamadas
‘vacunas’ contra el Alzheimer. Estas estaban formadas
por anticuerpos que se unen al péptido Aâ. Esta estrategia
fue transitoriamente abandonada al comprobarse en 2002 que este
tipo de tratamientos producía encefalitis (inflamación
del cerebro) en los pacientes que los recibían. Este resultado
no descarta la posibilidad de que el uso de anticuerpos pueda ser
modificado para evitar sus efectos colaterales de modo de convertirse
en un procedimiento eficaz para el tratamiento de la enfermedad.
Una alternativa a los anticuerpos es el uso de sustancias que, sin
serio, comparten con ellos la capacidad de combinarse con el péptido
Aâ, evitando su acción deletérea. En lo que
sigue se describirán algunos de los numerosos estudios que
emplearon este enfoque y que se dieron a conocer en el curso de
2006.
En 2004, científicos de la empresa Samaritan Pharmaceuticals
Inc. y de la Universidad de Georgetown de Estados Unidos informaron
en un artículo publicado en la revista Steroids que compuestos
derivados de intermediarios de la síntesis de esteroides
(los esteroides incluyen sustancias como el colesterol, los andrógenos
y estrógenos, y las hormonas de la corteza suprarrenal) se
unen al péptido Aâ evitando sus efectos sobre las neuronas.
Los estudios continuaron y así llevaron al desarrollo de
una droga llamada Caprospinol®. En diciembre de 2006 Samaritan
Inc. anunció que la Food and Drug Administration de los Estados
Unidos había autorizado el inicio de los ensayos clínicos
del Caprospinol®. Previamente se había demostrado que,
en animales transgénicos, el Caprospinol® detenía
la formación de placas amiloides y eliminaba las ya preexistentes
indicando que la droga bloqueaba las transiciones del péptido
Aâ.
Por su parte, investigadores de la empresa canadiense Neurochem
Inc., y del Departamento de Química de la Queen’s University
de Kingston, Ontario, publicaron en el número del 6 de noviembre
de 2006 de la revista Neurobiology of Aging estudios en los que
se demostró que una sustancia llamada Tramiprosate (ácido
3-amino-1-propansulfónico) que está registrada como
Alzhemed® también se asocia al péptido Aâ,
impidiendo que éste se una a sustancias llamadas glicosaminoglicanos
sulfatados (GAGs) y bloqueando así sus efectos. En el momento
de escribir esta nota (enero de 2007), la empresa estaba realizando
un ensayo clínico de fase III de esta droga (la fase III
es la última fase de las que preceden a la autorización
del uso de un medicamento).
Efectos similares a los de las drogas ya mencionadas fueron encontrados
en la escuela de medicina de la New York University: los resultados
fueron publicados el 20 de noviembre de 2006 en los Proceedings
of the National Academy of Sciences de EEUU y mostraron que un péptido
que tiene la misma secuencia de aminoácidos que un segmento
del péptido Aâ inhibe la formación de la placa
amiloide al impedir la combinación con el péptido
Aâ de un compuesto llamado apolipoproteína E que es
necesaria para que el péptido Aâ, se asocie y precipite.
En la revista Nature Medicine del 11 de junio de 2006 científicos
de la Universidad de Toronto informaron que una sustancia llamada
ciclohexanehexol antagoniza la unión del fosfatidil inositol
al péptido Aâ y protege o revierte las manifestaciones
del Alzheimer en ratones transgénicos.
Mejoramiento de la función sináptica
Una de las características de la enfermedad de Alzheimer
es el deterioro de la función de las sinapsis (ver recuadro
‘Sinapsis, neurotransmisores y receptores’) con el consecuente
déficit funcional de la compleja red neuronal necesaria para
la correcta actividad cerebral. Entre los distintos tipos de sinapsis
afectadas por la enfermedad, se destacan las que utilizan acetilcolina
como neurotransmisor. Estas sinapsis desempeñan un papel
central en los procesos de aprendizaje, memoria y en el procesamiento
de las emociones. Es por eso que los medicamentos actualmente utilizados
para tratar la enfermedad (Donepezilo, Rivastigmina y Galantamina)
están diseñados para aumentar la concentración
cerebral de acetilcolina. Sin embargo, la eficacia de estos medicamentos
es baja, ninguno de ellos proporciona más que un alivio parcial
y pasajero de los síntomas en las etapas iniciales e intermedias
de la enfermedad, y ninguno revierte las lesiones que ésta
provoca ni evitan su evolución fatal. De hecho, en noviembre
de 2006, el National Institute for Health and Clinical Excellence,
cuya misión es definir qué medicamentos pueden suministrarse
sin cargo en el sistema de salud del Reino Unido, recomendó
no suministrar estas drogas a nuevos pacientes y reducir las ya
usadas a pacientes con formas iniciales o intermedias de la enfermedad.
(Cabe consignar que, en enero de 2007, la empresa Eisai, que tiene
la licencia del Donepezilo, con el apoyo de Pfizer, que la comercializa,
recurrió a la justicia del Reino Unido para que esta decisión
se revirtiera).
En un intento de superar las deficiencias de los actuales medicamentos,
investigadores de la Universidad de California ensayaron mejorar
la actividad sináptica aumentando la sensibilidad de los
receptores de la acetilcolina. Para ello utilizaron un compuesto
llamado AF267B que facilita la acción de la acetilcolina
sobre la variedad de receptor muscarínico (ver recuadro ‘Sinapsis,
neurotransmisores y receptores’) para la acetilcolina que
abunda en las regiones del cerebro más afectadas en el mal
de Alzheimer. Los resultados de estos experimentos, que fueron publicados
en el número del primero de marzo de 2006 de la revista Neuron,
señalaron que el compuesto AF267B revirtió el déficit
en la capacidad de aprendizaje e hizo desaparecer las placas amiloides
y los ovillos neurofibrilares en las regiones más afectadas
del cerebro de los ratones transgénicos.
Además, en el curso de enero de 2007 varios laboratorios
estaban realizando ensayos clínicos con sustancias que aumentaban
la sensibilidad de otras variedades de receptores para la acetilcolina.
Sinapsis,
neurotransmisores y receptores
En cada neurona el impulso
nervioso consiste en una señal eléctrica -llamada
potencial de acción- que viaja con una amplitud y velocidad
constante. Este impulso pasa de una neurona a otra merced
a la liberación de sustancias llamadas neurotransmisoras
en una región especializada, llamada sinapsis. Una
vez que es liberado el neurotransmisor, este se difunde a
través del muy estrecho espacio que separa las neuronas
que participan en la sinapsis y al llegar a la que recibe
la señal se une a una proteína específica
llamada receptor. Esta unión desencadena la respuesta
de la neurona que recibió la señal permitiendo
así que la información contenida en el impulso
nervioso pase de una neurona a otra. La acetilcolina, uno
de los muchos neurotransmisores que participan en la función
cerebral, actúa sobre dos tipos de receptores: los
nicotinicos, que al unir acetilcolina abren un canal que permite
el paso de iones a través de la membrana, y los muscarínicos,
en los que la unión de la acetilcolina desencadena
una respuesta metabólica.
<
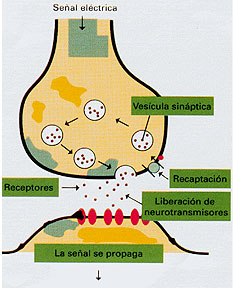
Figura 3. Esquema de una sinapsis. Se muestra solo la zona
de contacto entre dos neuronas. El impulso nervioso llega
en forma de una señal eléctrica hasta el extremo
de una neurona (arriba) donde el neurotransmisor está
contenido en las vesículas sinápticas. La llegada
del impulso nervioso desencadena un proceso que determina
que las vesículas
se unan a la membrana de la neurona y se abran al estrecho
espacio que separa ambas neuronas. liberando en este neurotransmisor
el que difunde hasta combinarse con los receptores de la otra
neurona que constituye la sinapsis (abajo). Una vez producida
la combinación la neurona se activa de un modo que
depende de ella y del neurotransmisor. Es así como
la información del impulso pasa de una neurona a otra.
Después de actuar el neurotransmisor puede ser destruido
o recaptado por la neurona que lo liberó para participar
en una nueva activación de la sinapsis. Nótese
que debido a que el neurotransmisor se libera solo desde una
de las neuronas y los receptores están solo en la otra,
en cada sinapsis el impulso nervioso pasa en una sola dirección. |
Aumento del nivel de la enzima Uch-L
1
Un planteo diferente a los anteriores fue utilizado por científicos
de la Universidad de Columbia en Nueva York. Consistió en
reestablecer los niveles de Uch-L 1 mediante la administración
de exógeno I de esta enzima. Los experimentos, que fueron
publicados el 25 de agosto de 2006 en la revista Cell, mostraron
que el suministro de Uch-L 1 corrige los déficit en la memoria
y en el aprendizaje de los ratones transgénicos. Los investigadores
interpretaron esto como una indicación de que la administración
de Uch-L 1 revierte las alteraciones que el Alzheimer provoca en
los complejos mecanismos bioquímicos que participan en la
memoria y en el aprendizaje.
Perspectivas
Los hallazgos aquí comentados deben ser confirmados con estudios
en seres humanos dado que no es predecible que los efectos favorables
observados en los ratones transgénicos se reflejen en la
mucha más compleja estructura y función cerebral de
los seres humanos.
A pesar de eso, los hallazgos permiten abrigar la esperanza de que
en un futuro no muy lejano se desarrollen medicamentos capaces de
evitar la aparición o revertir las lesiones de la enfermedad
de Alzheimer.
Si eso se lograra, millones prolongarían su vida útil
y, seguramente, nunca olvidarían el día en que recobraron
sus recuerdos.
Lecturas sugeridas
ARANCIO O, CAO Z, GONG B, MOOLMAN
D, LlU S, SHELANSKI M, STANISZÉWSKI A, VITOLO OV, ZHANG H
y ZHENG P, 2006, ‘Ubiquitin Hydrolase Uch-L 1 Rescues ì-Amyloid-lnduced
Decreases in Synaptic Function and Contextual Memory’, Cell,
126: 775-788.
BlLLINGS LM, CACCAMO A, FISHER A, GREEN KN, MARTINEZ-CORIA H, LAMERLA
FM y ODDO S, 2006, ‘M1 Receptors Play a Central Role in Modulating
AD-Iike Pathology in Transgenic Mice’, Neuron, 49, 671-682.
MEHTA PD, PANKIEWICZ J, PRELLI F, QUARTERMAIN D, SADOWSKI MJ, SCHOLTZOVA
H y WISNIEWSKI T, 2006, ‘Blocking the apolipoprotein E/amyloid-{beta}
interaction as A a potential therapeutic approach for Alzheimer’s
disease’, Proceedings of the National Academy of Sciences,
103:18787-18792. Este artículo está accesible en la
web.
SELKOE DJ, 2003, ‘Deciphering the genesis and fate of amyloid
â-protein yields novel therapies for Alzheimer disease’,
The Journal of Clinical Investigation, 110: 1375-1381.
Tanto Cell como Neuron están accesibles en la biblioteca
electrónica de la SECYT;
www.biblioteca.secyt.gov.ar/. Tanto el artículo de los Proceedings
of the National Academy of Sciences como el del The Journal of Clinical
lnvestigation están libremente accesibles buscando en www.pnas.org
y www.jci.orqg, respectivamente.
 Patricio
J Garrahan Patricio
J Garrahan
Profesor titular emérito. UBA.
Investigador superior.
CONICET.
e - mail: garrahan@mail.retina.ar
|
|