El infarto agudo de miocardio
El Comité de Redacción de Acta Bioquímica Clínica Latinoamericana ha seleccionado este artículo publicado en CIENCIA HOY – Volumen 22 - Número 131 – febrero - marzo 2013, pp. 31-35, para su difusión a través de FABAInforma
¿De qué se trata?
El infarto agudo de miocardio constituye una urgencia médica que requiere atención inmediata. Paradójicamente, algunas de las terapias más exitosas para su tratamiento pueden generar una nueva lesión, similar a la original. ¿Cuáles son los mecanismos responsables de estas complicaciones? ¿Cómo se puede avanzar en el diseño de nuevas terapias?
|
Romina Hermann, Débora E. Vélez,
María Gabriela Marina Prendes, Alicia Varela
Facultad de Farmacia y Bioquímica, Instituto de Química y
Metabolismo del Fármaco (UBA-Conicet)
El camino hacia nuevas terapias para su tratamiento
La isquemia cardíaca es la falta, o bien la disminución, del flujo sanguíneo coronario que produce un desequilibrio entre el aporte y la demanda de sangre oxigenada por el corazón. Este desequilibrio compromete tanto la entrega de oxígeno y nutrientes a las células como la remoción de sus desechos metabólicos, cuya acumulación resulta potencialmente tóxica. Si la intensidad y duración de la isquemia son suficientes para producir la muerte de las células cardíacas afectadas, ocurre el infarto agudo de miocardio (IAM). En cuestión de segundos cesa la actividad contráctil y se alteran las propiedades eléctricas en la zona afectada.
Las células que mueren por necrosis sufren cambios morfológicos que incluyen aumento de su volumen debido a la acumulación de metabolitos celulares durante la isquemia, que promueven la entrada de agua a la célula. También ocurre la ruptura de la membrana plasmática que rodea a la célula y la consecuente liberación del contenido celular que resulta, una vez restaurada la irrigación coronaria, en un proceso inflamatorio que también daña a las células vecinas. Entre las sustancias liberadas, la enzima creatina kinasa-isoenzima muscular cardíaca (CK-MB) y las proteínas troponina-T y troponina-I son usadas en la clínica como marcadores biológicos del daño sufrido por el músculo cardíaco (miocardio). El tejido necrótico es luego reemplazado paulatinamente por una matriz de colágeno que constituye el tejido cicatricial carente de propiedades contráctiles, es decir, de tejido no funcionante.
Según el Informe sobre la salud en el mundo 2008 de la Organización Mundial de la Salud, el proceso de urbanización, el envejecimiento y los nuevos modos de vida en todo el mundo están haciendo que las enfermedades cardiovasculares sean una causa cada vez más importante de morbilidad y mortalidad. Al respecto, las causas más comunes del IAM son la formación de un trombo sanguíneo, generalmente ocasionada por rotura o erosión de una placa de ateroma (depósito e infiltración de lípidos, células inflamatorias, macrófagos en las paredes de las arterias), lo cual produce la obstrucción del vaso coronario e isquemia de la zona del miocardio irrigada por la arteria afectada (figura 1).
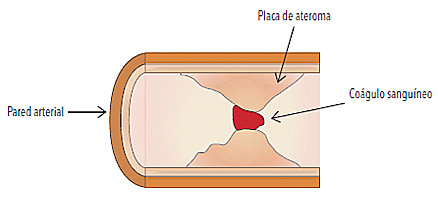
• Figura 1. Representación esquemática en corte transversal del bloqueo de una arteria coronaria provocado por un trombo sanguíneo.
El grado de extensión de la necrosis depende de la extensión del área en riesgo, que es la región irrigada por la arteria afectada, de la existencia de circulación colateral que permita cierto flujo de sangre en dicha región y de la duración de la isquemia. De ello se desprende la importancia de disminuir el tiempo que se tarda desde el comienzo de los síntomas hasta la restauración de la irrigación sanguínea del área de riesgo (reperfusión) para conseguir que la extensión de la necrosis celular, y por lo tanto el tamaño del infarto, se reduzca al mínimo.
La reperfusión, sea mediante la administración de medicamentos para disolver el trombo intravascular o mediante otros procedimientos como la angioplastia coronaria transluminal percutánea, es la intervención imprescindible para salvar al tejido isquémico de la muerte celular segura. La angioplastia consiste en la introducción de un balón en el extremo distal de un catéter frente a la oclusión, que se infla hasta mejorar el flujo sanguíneo. Esta intervención finaliza con la implantación de un dispositivo tubular (stent) que evita el colapso vascular luego de la dilatación del vaso.
Sin embargo, numerosas investigaciones han demostrado que, paradójicamente, puede ocurrir una nueva lesión por la reperfusión que contrarresta sus efectos beneficiosos y que suele ocurrir durante los minutos siguientes a la intervención. Por ello la reperfusión es considerada ‘un arma de doble filo’. Las causas de este fenómeno, conocido como daño o lesión por reperfusión, están siendo intensamente investigadas con la finalidad de mejorar la eficacia de las intervenciones empleadas para restablecer el riesgo sanguíneo mediante tratamientos cardioprotectores coadyuvantes. En los últimos años se ha producido un gran avance en la identificación de los factores que desempeñan un papel fundamental en la lesión por reperfusión, lo cual ha permitido nuevas dianas terapéuticas potencialmente útiles para prevenirla y limitar así el tamaño del infarto.
Uno de los mecanismos que actualmente está recibiendo gran atención es la pérdida de la estructura y función de las mitocondrias durante el proceso de isquemia y reperfusión. Estas organelas constituyen las centrales energéticas de la célula. Poseen dos membranas, una externa y otra interna, que delimitan tres compartimientos: el citosol, el espacio intermembrana y la matriz mitocondrial. La membrana interna posee una permeabilidad altamente selectiva y forma numerosos pliegues o crestas donde se asientan proteínas de transporte y enzimas que conforman la vía de la fosforilación oxidativa, responsable de la síntesis del trifosfato de adenosina (ATP) en presencia de oxígeno. El ATP es un compuesto fundamental ya que almacena gran cantidad de energía para las funciones biológicas vitales, que se libera cuando parte de los enlaces fosfato de la molécula se separan. Más del 95% del ATP, imprescindible para mantener la viabilidad celular y la adecuada funcionalidad cardíaca, se produce en las mitocondrias en presencia de oxígeno.
Bajo situaciones de isquemia seguida de reperfusión se produce un grave deterioro en la producción de energía. Durante la isquemia la disminución o falta de oxígeno que ocurre en la zona afectada del corazón ocasiona un rápido agotamiento de sus reservas energéticas, representadas principalmente por el ATP. De manera conjunta y gradual, durante la isquemia aumenta la acidez del contenido celular (la concentración de protones o iones hidrógeno), por lo que se produce el fenómeno de acidosis celular. La recuperación del balance normal de la concentración de protones resulta en una paulatina activación del intercambiador sodio-protón, proteína transportadora presente en la membrana que rodea la célula muscular cardíaca o miocito, que exporta protones aprovechando la energía que libera la entrada de sodio a la célula a favor de su gradiente electroquímico. Dicho gradiente depende de la actividad de una bomba presente en las membranas celulares, la bomba sodio-potasio ATPasa, que elimina sodio de la célula e introduce potasio, y consume para ello la energía proveniente del ATP. El exceso de protones generados durante el período de isquemia junto con la disminución de la actividad de la bomba sodio-potasio ATPasa producen un aumento del sodio intracelular. A su vez, este hecho afecta también el intercambiador sodio-calcio, cuya función es mantener baja la concentración de calcio intracelular al exportarlo aprovechando la energía que libera la entrada de sodio a favor de su gradiente electroquímico, contribuyendo de esta manera al aumento de calcio intracelular durante este período (figura 2).
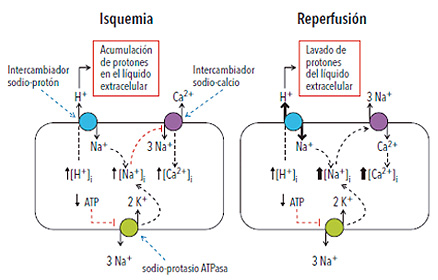
• Figura 2. Mecanismos que alteran la homeostasis iónica durante la isquemia y la reperfusión. El exceso de protones (H+) generados durante la isquemia es exportado hacia el medio extracelular en intercambio con sodio (Na+). Este proceso junto con la disminución de la actividad de la bomba sodio-potasio (K+) ATPasa producen un aumento del sodio intracelular, que a su vez genera una sobrecarga celular de calcio (Ca2+) vía el intercambiador sodio-calcio. Durante la reperfusión, la remoción de los protones del medio extracelular y la persistente acidosis intracelular producen hiperactivación del intercambiador sodio-protón, que acompañada por el agotamiento de ATP exacerba la sobrecarga celular de calcio.
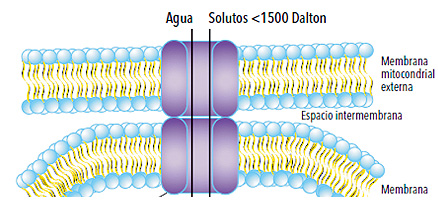
• Figura 3. El poro de transición de la permeabilidad mitocondrial. La apertura del poro de transición de permeabilidad mitocondrial (PTPM) –poro inespecífico de alta conductancia– produce un aumento de la permeabilidad de la membrana mitocondrial interna, permitiendo el paso de moléculas de hasta 1500 Dalton de peso molecular con la consiguiente entrada de agua. Por otra parte, convierte a la mitocondria de productora en consumidora de ATP y permite la liberación de factores apoptóticos.
Así, durante los primeros minutos de la reperfusión, y principalmente como consecuencia de la puesta en marcha de mecanismos dirigidos a corregir la acidosis intracelular, puede precipitarse un empeoramiento abrupto de la sobrecarga de sodio y calcio por hiperactivación de los intercambiadores sodio-protón y sodio-calcio, funcionando este último de manera revertida favoreciendo el ingreso de calcio a la célula. Al proceso de agotamiento de ATP y los desbalances de sodio y calcio ya descriptos (figura 2) se agrega la producción de radicales libres del oxígeno, moléculas muy pequeñas altamente reactivas, que aumentan marcadamente con el reingreso del oxígeno a la célula cardíaca y debido a su liberación por parte de células sanguíneas fagocíticas que intervienen en la respuesta inflamatoria aguda en los focos de necrosis tisular. La combinación de estos eventos durante los primeros minutos de la reperfusión puede desencadenar un aumento acentuado de la permeabilidad de la membrana mitocondrial interna de tal manera que ésta pierde su selectividad permitiendo el paso indiscriminado de moléculas de elevado peso molecular con la consiguiente entrada de agua a la mitocondria (figura 3). Esto genera un importante edema mitocondrial que puede a su vez producir la ruptura de la membrana mitocondrial externa (figura 4).
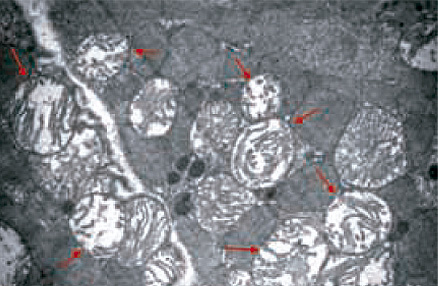
• Figura 4. Microscopia electrónica de una biopsia de miocardio de rata tomada a los 75 minutos de la reperfusión luego de un período de 75 minutos de isquemia. Se observan mitocondrias marcadamente hinchadas, edematizadas, con desorganización y separación de las crestas y aclaramiento de la matriz mitocondrial (flechas).
Esta alteración en la permeabilidad selectiva de la membrana mitocondrial interna parece deberse a la apertura de un poro inespecífico de alta conductancia, conocido como poro de transición de la permeabilidad mitocondrial (PTPM), que convierte a la mitocondria de productora en consumidora de ATP. Este brusco cambio, que produce un grave deterioro en la producción de energía, resulta incompatible con la supervivencia de la célula cardíaca y promueve la muerte celular aguda por necrosis. Cabe destacar que si bien este fenómeno ocurre durante los primeros minutos de la reperfusión, el escenario para ponerlo en marcha comienza a prepararse durante el período isquémico.
Por otra parte, a partir de su material genético cada célula posee un programa de muerte llamado apoptosis, que al ser desencadenado de manera espontánea o inducida conduce a la desaparición de la célula. La apoptosis ocurre fisiológicamente durante la embriogénesis, el desarrollo y mantenimiento normal de órganos y tejidos, la regulación del sistema inmunitario y durante el envejecimiento. La apertura del PTPM podría desencadenar este tipo de muerte celular programada a más largo plazo, ya que permite la liberación de factores que desencadenan la apoptosis. Con independencia del mecanismo exacto responsable de este proceso de transición de la permeabilidad mitocondrial, este evento puede marcar el punto de no retorno en el proceso de muerte celular (figura 5).
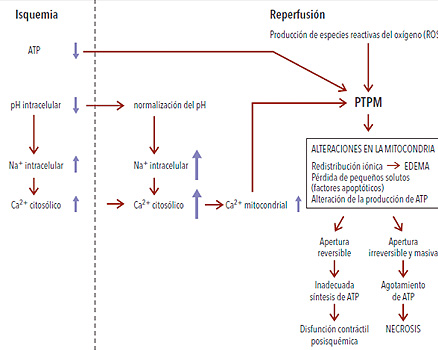
• Figura 5. Eventos que durante la isquemia y la reperfusión llevan a la apertura del PTPM. Si bien la apertura del PTPM ocurre durante los primeros minutos de la reperfusión, durante el período isquémico comienza a prepararse el escenario para que ello ocurra.
Se ha demostrado en animales de experimentación que la prevención de la apertura del PTPM protege al corazón del daño por isquemia-reperfusión, por lo que este poro se ha convertido en una importante diana para el desarrollo de estrategias farmacológicas cardioprotectoras, como la administración del agente inmunosupresor ciclosporina A (Cs A), droga que inhibe directamente la apertura del poro, o intervenciones que aminoren las causas que conducen a su apertura. En un ensayo clínico piloto llevado a cabo en Francia, y cuyos resultados se publicaron en 2008, la Cs A ha demostrado efectos beneficiosos en pacientes sometidos a revascularización mediante angioplastia. Sin embargo, la Cs A posee efectos colaterales, por lo que no representa el tratamiento coadyuvante ideal a ser usado en la reperfusión del miocardio.
En 1986, en el Duke University Medical Center de Carolina del Norte, Estados Unidos, descubrieron que cortos períodos de oclusión coronaria antes de una oclusión sostenida reducían el infarto de miocardio en un 70%. Este fenómeno, que se conoce como preacondicionamiento isquémico, desencadena mecanismos endógenos de cardioprotección contra el daño por isquemia-reperfusión, que son los más potentes descubiertos hasta el presente. Los mecanismos moleculares responsables del efecto cardioprotector del preacondicionamiento isquémico no han sido aún totalmente dilucidados, pero numerosas evidencias experimentales señalan al PTPM como efector final fundamental para la preservación de la funcionalidad mitocondrial y de la viabilidad celular. Se han descripto dos fases de protección: una fase inicial clásica o primera ventana de protección que actúa de una a dos horas luego de aplicados los cortos períodos de isquemia no letal (estímulo preacondicionante) previos a la isquemia sostenida y una fase tardía conocida como la segunda ventana de protección que se manifiesta veinticuatro horas después de dicho estímulo. La necesidad de aplicar el estímulo preacondicionante antes de la isquemia del miocardio, que en el caso del IAM es difícil de predecir, es un obstáculo para su empleo en el ámbito clínico. En 2003, investigadores de Atlanta, Estados Unidos, lograron una notable reducción del tamaño del infarto aplicando en animales de experimentación esa estrategia cardioprotectora en el inicio de la reperfusión. A este fenómeno se lo denominó posacondicionamiento isquémico. Numerosos estudios experimentales señalan al PTPM como efector final de los mecanismos moleculares endógenos disparados por el posacondicionamiento isquémico. Estudios experimentales de laboratorio demostraron que las mitocondrias aisladas del área de riesgo de corazones pre y posacondicionados sometidos a isquemia-reperfusión muestran una morfología altamente preservada, mayor resistencia a la apertura del PTPM frente a una sobrecarga de calcio y, por supuesto, una marcada reducción del tamaño del tejido infartado. Dos años después de las investigaciones realizadas en Atlanta, el posacondicionamiento isquémico fue aplicado por primera vez y con éxito en un ensayo clínico llevado a cabo en Francia, en pacientes con IAM sometidos a angioplastia coronaria. Se empleó un protocolo en el cual el balón de angioplastia fue inflado y desinflado cuatro veces durante un minuto, dentro del primer minuto de la reperfusión coronaria. La reducción del tamaño del infarto, evaluado mediante la determinación de los niveles séricos de CK-MB durante las 72 horas posteriores a la intervención, en un 36% con respecto a pacientes no sometidos a posacondicionamiento aporta un fuerte sustento a la existencia de la lesión por reperfusión. Sin embargo, ensayos posteriores arrojaron resultados discordantes, por lo que es aún necesario profundizar en el conocimiento del papel que juegan en estas intervenciones factores como la edad, el género o la existencia de enfermedades como la diabetes, la hipertensión arterial, la hiperlipidemia, el síndrome metabólico, la hipertrofia ventricular, etcétera. También es necesaria la realización de ensayos clínicos multicéntricos y a gran escala para confirmar en el ámbito clínico los efectos beneficiosos de esta estrategia que ha arrojado resultados positivos en el laboratorio. Finalmente, la posibilidad de salvar el miocardio mediante tratamientos farmacológicos coadyuvantes aplicados durante la reperfusión coronaria representa una nueva oportunidad terapéutica para los pacientes con IAM. No obstante, resta aún un largo camino por recorrer. A pesar de las numerosas evidencias del papel desempeñado por el PTPM en el daño por isquemia-reperfusión y en los efectos beneficiosos del pre y del posacondicionamiento isquémicos, falta todavía establecer una relación causal fehaciente entre la inhibición de la apertura del poro y los efectos cardioprotectores de estas intervenciones. Un obstáculo importante que se interpone para el logro de este objetivo es la incertidumbre acerca de la exacta estructura molecular del PTPM y de los efectos colaterales de las herramientas farmacológicas con que se cuenta para inhibir su apertura, que no impiden descartar otras dianas moleculares en sus mecanismos de acción. Además, la dilucidación de su estructura y su papel en situaciones de isquemia trasciende el IAM, ya que el PTPM parece también desempeñar un papel clave en los daños causados en el accidente cerebro-vascular. En resumen, el IAM constituye una urgencia médica por definición y se debe buscar atención profesional inmediata. Las demoras son un error grave que se cobra miles de vidas cada año. El pronóstico vital de un paciente con IAM depende de la extensión de la lesión y del tiempo transcurrido hasta la atención médica. Sin embargo, la reperfusión del miocardio puede llevar aparejada una nueva lesión que amortigua sus efectos beneficiosos originales. Por ello, el diseño de estrategias cardioprotectoras novedosas que sean efectivas aun en presencia de otras enfermedades representa un importante requerimiento para mejorar los resultados clínicos y un desafío para la ciencia en el área biomédica.
Lecturas sugeridas
• CINGOLANI HE, PÉREZ NG y CAMILIÓN DE HURTADO MC, 2000, ‘¿De qué es culpable el intercambiador Na+/H+ en cardiología?’, Medicina, vol. 60, pp. 709-721.
• MOSCA SM, ‘Pre y posacondicionamiento isquémicos: papel del poro de permeabilidad transitoria de la mitocondria’, Revista de la Federación Argentina de Cardiología 2011; vol. 40 (2), pp. 115-120.
• Romina Hermann, Bioquímica, Facultad de Farmacia y Bioquímica, UBA. Ayudante de primera, FFYB, UBA. Becaria de doctorado, UBA rhermann@ffyb.uba.ar
• María Gabriela Marina Prendes, Doctora en Bioquímica, UBA. Jefe de trabajos prácticos, FFYB, UBA. gmarina@ffyb.uba.ar
• Alicia Varela, Doctora en Bioquímica, UBA. Profesora adjunta, FFYB, UBA. avarela@ffyb.uba.ar
• Débora Elisabet Vélez, Bioquímica, FFYB, UBA. Ayudante de segunda, FFYB, UBA. Becaria estímulo, UBA.
velez.debora@hotmail.com
|
|