| El
Comité de Redacción de Acta Bioquímica Clínica
Latinoamericana ha seleccionado este artículo publicado en la revista
“Diagnóstico Bioquímico y Molecular”, N.º
5, 2005, para su difusión a través de FABA Informa.
¿A qué se llama "-talasemia
?
La "-talasemia es un trastorno hereditario que afecta la síntesis
de las cadenas de la hemoglobina (Hb) y por consecuencia, el transporte
de oxígeno a todos los tejidos. Esta hemoglobinopatía incluye
diferentes formas de anemia. La gravedad y el tipo de la misma dependen
del defecto genético que la origina.
Estructura de la molécula de hemoglobina
 La Hemoglobina
es una proteína constituida por 4 cadenas proteicas, 2 cadenas
alfa y 2 no alfa (beta, delta o gamma) unidas a un grupo hemo. Las "-talasemias
se producen por alteraciones en la síntesis total o parcial, de
cadenas alfa. La Hemoglobina
es una proteína constituida por 4 cadenas proteicas, 2 cadenas
alfa y 2 no alfa (beta, delta o gamma) unidas a un grupo hemo. Las "-talasemias
se producen por alteraciones en la síntesis total o parcial, de
cadenas alfa.
Introducción
Las hemoglobinopatías constituyen un conjunto de anemias hemolíticas
que podemos subdividir en tres grandes grupos:
Hemoglobinopatías cualitativas o estructurales: originadas por
hemoglobinas anómalas que por distintos mecanismos desencadenan
crisis hemolíticas.
Hemoglobinopatías cuantitativas o Talasemias: producidas por hemoglobinas
normales pero donde está alterada la síntesis de una de
las cadenas de la hemoglobina.
De acuerdo a la cadena mutada las talasemias se clasifican en:
"-talasemia
$-talasemia
$*-talasemia
$*(-talasemia
Hemoglobinopatías talasémicas: donde las hemoglobinas formadas
son anormales y su síntesis está disminuida.
En este informe nos ocuparemos específicamente de la "-talasemia
de menor frecuencia en nuestra población.
La "-talasemia se produce por mutaciones en los genes que codifican
para las cadenas de "-globina de la molécula de Hemoglobina.
Esta alteración desencadena cuadros de distinta severidad que podrían
pasar inadvertidos o ser totalmente incompatibles con la vida.
La mayor frecuencia de genes "-talasémicos se localiza en
el sudeste asiático y sobre el oeste de África. En Thailandia,
la incidencia se encuentra entre el 4,8 y el 10%, alrededor de 250.000
personas tienen alguno de los síndromes "-talasémicos.
Con menor frecuencia aparece en India, Kuwait, Grecia e Italia. Si bien
en nuestra población, de ascendencia predominantemente mediterránea,
no es tan frecuente como la $-talasemia, es probable que su verdadera
incidencia resulte subestimada debido a la dificultad que representa llegar
a su diagnóstico.
El objetivo de esta comunicación es difundir breves nociones sobre:
• sus principales
bases moleculares
• su clasificación
• su fisiopatología
• y facilitar su diagnóstico
a través del laboratorio de rutina.
Principales aspectos moleculares
Cada tipo de cadena (" o $) se encuentra codificada por un gen distinto.
Las cadenas de la "-globina están codificadas por genes localizados
en el cromosoma 16 en el "cluster de genes alfa". Cada cromosoma
16 tiene dos genes -"2 y "1- idénticos que rigen la síntesis
de cadenas alfa, por lo que la dotación genética normal
es ""/"". El "cluster de genes no alfa",
que contiene los genes que codifican para las cadenas de $, * y (-globina,
se localiza en el cromosoma 11.
A diferencia de las $-talasemias, la mayoría de las "-talasemias
se producen por deleciones que involucran la pérdida de miles de
pares de bases (pb) del cromosoma:
"+-talasemia (-"): la deleción determina la pérdida
de una de las dos copias de los genes alfa.
" 0-talasemia (--): la deleción abarca los dos genes alfa
del cromosoma.
De acuerdo a la combinación de estas deleciones se observan los
siguientes genotipos " -talasémicos:
-"/"" ("-talasemia-2): una copia del gen delecionada.
--/"" ("-talasemia-1): dos copias delecionadas en el mismo
cromosoma (deleción cis).
-"/-" (" -talasemia-1): una copia delecionada en cada cromosoma
(deleción trans).
--/-" (Enfermedad de la Hb H): deleción de tres copias.
--/-- (Síndrome de Hidropesía Fetal con Hb Bart), deleción
de cuatro copias.
Las deleciones más frecuentes son:
--"3,7: es una deleción de 3,7 Kpb, también se conoce
como rightward deletion; se pierde parte del gen "2 y parte del gen
"1 quedando un gen alfa de fusión; es la más frecuente
en el Mediterráneo y Africa.
-- "4,2: es una deleción de 4,2 Kpb también llamada
leftward deletion; se pierde el gen "2 completo; es la más
frecuente en el sudeste asiático.
Existen deleciones que incluyen la pérdida de los 2 genes ":
--MED: deleción de más de 26,5 Kpb.
-(")20,5: deleción de 20,5 Kpb.
Las mutaciones no delecionales son menos frecuentes, se observan generalmente
en el gen "2 ("T"), u ocasionalmente en el gen "1
(""T).
Clasificación
La "-talasemia pertenece al grupo de las hemoglobinopatías
cuantitativas; se producen por disminución o ausencia de síntesis
de las cadenas alfa, pero las cadenas sintetizadas son estructuralmente
normales a diferencia de lo que ocurre en las hemoglobinopatías
cualitativas como la Drepanocitosis donde se sintetizan cadenas estructuralmente
anómalas en cantidad normal.
El fenotipo eritrocitario y la clínica dependerán de la
gravedad de la alteración genética: la deleción de
un solo gen " no se acompaña de alteraciones clínicas,
mientras que la deleción de los cuatro genes " provoca la
muerte in utero.
De acuerdo a que la disminución o ausencia sea de uno, dos, tres
o cuatro genes, tendremos los siguientes cuadros hematológicos:
"-talasemia silente ("-talasemia-2): si la deleción ha
afectado un solo gen no se produce alteración clínica alguna.
La única manifestación del trastorno genético será
un hemograma con una cifra de hematíes en la zona alta de la normalidad
y un VCM algo disminuido. La amplitud de distribución eritrocitaria
(ADE) es normal. El genotipo es -"/"".
Rasgo talasémico ("-talasemia-1): puede tener dos genotipos
distintos (cis: --/"" o trans: -"/-"). Las manifestaciones
clínicas son mínimas o nulas y en el hemograma aparece una
anemia moderada con microcitosis y poliglobulia. La hiperferritinemia
es infrecuente, por lo que la concentración elevada de ferritina
debe hacer sospechar la presencia concomitante de una hepatopatía
o de hemocromatosis. La prevalencia de estas dos formas de "-talasemia
en España alcanza el 0,02-0,5%. Se debe establecer el diagnóstico
diferencial con la anemia ferropénica (en la que rara vez la cifra
de hematíes es tan alta y la ADE suele ser superior a la normal)
y con otros tipos de talasemias (básicamente $-talasemia, que cursa
con aumento de Hb A2 o la *$-talasemia, en la que aumenta la Hb F).
Enfermedad por Hb H: con deleción de tres genes alfa (genotipo
--/-"). Es frecuente en China e Indonesia y se han descripto también
algunos casos en Italia, Sudamérica y en España. Cursan
con un cuadro clínico de anemia hemolítica de intensidad
moderada y moderada esplenomegalia. Si bien este cuadro podría
compararse con una $-talasemia homocigota su gravedad no es tan significativa
y esto es fácilmente justificable por la fisiopatología
de la anemia ya que como mencionáramos anteriormente el componente
de eritropoyesis ineficaz (si bien existe) no es tan acentuado debido
a la presencia de las Hemoglobinas H y Bart.
Síndrome de Hidropesía Fetal con Hb Bart: la deleción
de los cuatro genes alfa (--/--), es incompatible con la Vida.
Produce en el feto un grave cuadro de hidropesía secundaria a la
intensa anemia, con gran hepato-esplenomegalia, que causa la muerte fetal
al final del embarazo o pocas horas después del parto. No se ha
descripto en España ni en Sudamérica.
(ver cuadro)
Fisiopatología y cuadro clínico
La disminución de la síntesis de cadenas alfa provocará
un desbalance de cadenas alfa / no alfa; en el feto y el recién
nacido existirá un exceso de cadenas gamma y en el adulto, un exceso
de cadenas beta. En el feto se producen tetrámeros de cadenas gamma
((4) la Hb. Bart, que tiene elevada afinidad por el oxígeno. El
exceso de cadenas beta produce la Hb. H (tetrámeros $4), que es
inestable e induce lisis de los hematíes.
¿Cuáles son las consecuencias de la formación de
estos tetrámeros?
Los tetrámeros, a pesar de ser inestables no precipitan en la médula
ósea, por lo tanto la eritropoyesis es más efectiva en estas
anemias que en la $-talasemia (donde se producen acúmulos de cadenas
" que precipitan en médula). La inestabilidad de la Hb H origina
su precipitación en el glóbulo rojo. Los cuerpos de inclusión
producidos son atrapados por el bazo provocando la disminución
de la vida media del eritrocito. Al mismo tiempo, tanto la Hb Bart como
la Hb H, al no poseer cadenas alfa, poseen alta afinidad por el oxígeno
(no hay interacción hem-hem) y la curva de disociación del
oxígeno se asemeja a la de la mioglobina. Por esto, las formas
severas de "-talasemia se deben a la producción deficiente
de Hb la formación de homotetrámeros y el componente hemolítico
debido a su precipitación en los glóbulos rojos.
Contribución del laboratorio de rutina en el diagnóstico
de la anemia
Recuento de sanqre entera:
Se indica en todos los individuos en los que se sospecha un desorden en
la síntesis de cadenas de globina. Los índices hematimétricos
son de particular importancia en el screening de " y $-talasemias.
En las talasemias el VCM es inferior a 80 f1 Y el HCM es menor de 27 pg,
en particular para la " 0-talasemia (enfermedad de Hb H) el valor
de HCM es menor a 25 pg. El extendido de sangre periférica es también
útil, pudiéndose observar anisocitosis, con microcitosis
y anisocromía con hipocromía. El recuento de reticulocitos
es importante como indicador de estado hemolítico como en el caso
de la enfermedad de la Hb H. De todas formas siempre deben realizarse
pruebas de confirmación más allá de los resultados
que arrojen los índices hematimétricos y el extendido de
sangre periférica.
Electroforesis de Hemoqlobina en acetato de celulosa, pH
alcalino:
Los portadores de "-talasemia y el rasgo "-talasémico
presentan un cuadro electroforético normal. La Enfermedad de Hb
H presenta en el adulto una banda de Hb H ($4) de movilidad rápida
de entre 5 a 30% de concentración. Esta Hb es bastante lábil
por lo que se sugiere que la electroforesis sea realizada el mismo día
de la extracción de la muestra para evitar su pérdida y
una corrida electroforética falsamente normal. En el momento del
nacimiento se presenta también una banda de Hb Bart ((4) de 10
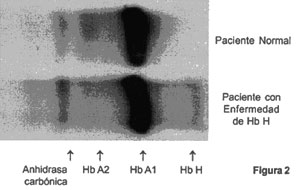
a 25%. A continuación se presenta una corrida electroforética
con presencia de Hb H: (Ver imagen 1)
Imagen1
Cuerpos de inclusión eritrocitarios:
Las inclusiones eritrocitarias más importantes que aparecen en
las hemoglobinopatías son las inclusiones de Hb H (precipitados
de tetrámeros de cadenas $) que se encuentran en "-talasemia.
Para la demostración de estas inclusiones se utiliza azul de metileno
o azul brillante de cresilo al 1%. La técnica es similar a la que
se realiza para la determinación de reticulocitos, incubándose
a 37 °C por 1 hora, luego se realizan extendidos donde se pueden observar
inclusiones esféricas color verde oscuro en los eritrocitos. En
la "+-talasemia sólo se observan ocasionalmente inclusiones
de Hb H (1:1000 a 1:10000), por lo que su búsqueda debe ser exhaustiva,
aproximadamente unos 15 minutos en cada frotis. En la enfermedad de la
Hb H son más numerosos, encontrándose en más del
30% de las células rojas. (Ver imágenes 2 y 3)
Imagen 2 y 3
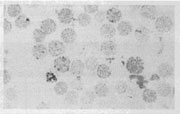 2-
Clinical Hematology 2-
Clinical Hematology
Víctor Hoffbrand
John E. Petit
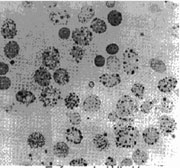 3-
Enfermedad Hb. H 3-
Enfermedad Hb. H
Practical Haematology
Sir John V. Dacie. S.M.Lewis
Tests de inestabilidad
La Hb H tiene una inestabilidad intermedia entre la Hb A normal y las
hemoglobinas típicamente inestables. Existen varios métodos
disponibles para la demostración de la inestabilidad de la Hb:
el test de n-butanol, de isopropanol y la prueba de inestabilidad al calor.
Como resumen podemos concluir las siguientes observaciones:
Portador sílente "
Microcitosis borderlíne y un HCM normal o apenas disminuido, corrida
electroforética de Hb normal. El diagnóstico definitivo
sólo puede hacerse por técnicas de biología molecular.
Rasgo talasémico "
Anemia leve, microcitosis persistente, ferremia y ferritina dentro de
los parámetros normales, inclusiones eritrocitarias de Hb H (1
en 105 glob. rojos). Se observa una relación de síntesis
de cadenas "/$ disminuida (aproximadamente 0,6).
Enfermedad de Hb H
Anemia hemolítica crónica con el cuadro clínico típico
de una talasemia intermedia. VCM y HCM por debajo de los limites normales.
En frotis se observan: anisopoiquilocitosis, hipocromía, target
cells y 4-5% de reticulocitos. Las inclusiones eritrocitarias de Hb H
superan el 30%.
Síndrome de Hídropesía Fetal con Hb Bart
Ausencia total de cadenas ", lo que es incompatible con la vida.
Los mortinatos presentan edema grave, anemia marcada y hepatoesplenomegalia.
Se detectan grandes cantidades de Hb, Bart ((4) y algo de Hb H ($4), que
se pueden visualizar por electroforesis.
Caso clínico
Paciente de sexo femenino y de 20 años de edad (ver cuadro aparte)
Frotis: intensa anisocitosis, anisocromía, microcitos, hipocromía,
target cells, poiquilocitosis, eliptocitos
Ferremia: 74 mg/dl
Corrida electroforética: Hb A2: 1,9% Hb F: 0,9% Hb H: 5% Hb A:
92,2% ( Figura 2)
Cuerpos de inclusión de Hb H: 70%
Test isopropanol: 10 min; normal: 25 min
Estudio molecular: genotipo --/- " determinado por Southern Blot
Conclusión: la combinación de las distintas determinaciones
permitió confirmar la presencia de Enfermedad de Hb. H.

Consejo genético a futuros padres antes y durante el embarazo
Entre los desórdenes que deben predecirse en los futuros padres
está la "°-talasemia o la enfermedad de Hb H y en el feto
la presencia del Síndrome de hidropesía fetal.
El diagnóstico prenatal para hemoglobinopatías ha progresado
con el avance de las técnicas de obtención y análisis
de muestras. El ADN fetal se obtiene a partir de una muestra de vellosidades
coriónicas de semana 10-12 de gestación y las mutaciones
del gen alfa se identifican en forma directa a través de técnicas
de biología molecular. El screening inicial para "°-talasemia
se basa en el análisis de los índices hematimétricos.
Se requiere una investigación más profunda cuando el HCM
es < 25 pg y $ y *$-talasemia y Hb Lepore han sido descartadas a través
de una electroforesis de Hb. Cuando se sospecha que una mujer es portadora
de "°-talasemia se recomienda sólo proceder al análisis
molecular si su pareja también tiene un HCM < 25 pg. La presencia
de $-talasemia no excluye la coexistencia de "°-talasemia (ya
que estas alteraciones al provenir de distintos cromosomas, se heredan
en forma independiente) y la falla en poder detectarlo puede conducir
a la concepción de un feto con Hb Bart y fetos hidrópicos
.
El screening para "°-talasemia (fenotipo -/"") debe
realizarse en mujeres de los siguientes orígenes étnicos:
China, sudeste asiático, Grecia, Turquía y Chipre. Si existe
consanguinidad entre la mujer y su pareja las indicaciones del screening
son aún mayores. En mujeres de origen indio, africano y afro-caribeño
no es necesario realizarlo, ya que generalmente proviene del genotipo
–"/-".
Un valor de Hb A2 en el límite superior normal en un paciente con
índices hematimétricos compatibles con talasemia puede provenir
de la coexistencia de " y $ talasemia.
No es necesario testear a mujeres originarias del norte de Europa ya que
la "°-talasemia no aparece en este grupo étnico.
Screening basado en el hallazgo de microcitosis
Frente al hallazgo de una microcitois, en primera instancia, se debe descartar
un déficit de Hierro (Fe), que es la causa más frecuente
de anemia.
Si la microcitosis persiste luego de normalizar los parámetros
de Fe, se debe comenzar con el estudio de las talasemias.
Recurrimos entonces a la electroforesis de Hb; si en el patrón
electroforético aparece una banda de Hb A2 aumentada se sugiere
el diagnóstico de una " Talasemia heterocigoto.
Si por el contrario, el cuadro electroforético es normal debe considerarse
el diagnóstico de " Talasemia.
Si el paciente está en edad reproductiva, pertenece a un grupo
étnico apropiado y su HCM es < 25 pg, debe considerarse un test
definitivo para "°-talasemia. Si el paciente no pertenece a un
grupo étnico de riesgo y su HCM es > 25 pg, no se recomienda
seguir investigando. En grupos étnicos donde la "°-talasemia
es más frecuente, la presencia de índices talasémicos
con Hb A2 normal puede deberse a la coexistencia de $ o *-talasemia. El
diagnóstico debe confirmarse con estudios de síntesis de
cadenas de globina o de diagnóstico molecular.
Tratamiento de la "-talasemia:
En el tratamiento de la "-talasemia se deben tener en cuenta los
siguientes aspectos:
• La edad, estado
general de salud y antecedentes médicos del paciente
• La gravedad de la
enfermedad
• La tolerancia del
paciente a determinados medicamentos, procedimientos o terapias
El tratamiento para la "-talasemia puede incluir:
• Dosis diarias de
ácido fólico
• Extirpación
quirúrgica del bazo (si fuera necesario)
• Transfusiones de
sangre (según sea necesario)
• Quelantes de hierro
Conclusión
Debido a la posible subestimación de aquellas "-talasemias
con deleción de uno o dos genes alfa, que son indetectables electroforéticamente
se sugiere el estudio de todas aquellas microcitosis con VCM entre 70
y 80 fl e hipocromias menores a 27% de HCM, con electroforesis normal
y niveles de hierro normales.
Para ello desde el laboratorio de rutina se debe recurrir al hemograma,
electroforesis de Hb, cuerpos de Heinz y si todo esto es normal, se aconseja
el diagnóstico molecular para identificar las posibles mutaciones
responsables del cuadro hematológico.
BIBLlOGRAFÍA
1. Dacie JV, Lewis SM. Practical Haematology. Eight Edition.
Churchil Livingstone; 1995.
2. Sans-Sabrafen J, Besses Raebel C, Vives Corrons JL,
Hematología Clínica, Cuarta Edición. Ediciones Harcourt,
S. A.; 2001.
3. Richard Lee G, Bithell TC, Foerster J, Athens JW,
Lukens JN. Wintrobe's. Clinical Hematology. Ninth Edition. Lea & Febiger;
1993.
4. Hoffbrand V, Petit JE. Color Atlas of Clinical Hematology;
Third Edition. Editorial Mosby; 2000.
5. Weatheral DC, Clegg JB - The Thalassemia Syndromes.
3rd. edition; Oxford: Blackwell Scientific; 1981.
6. D' Alton MA, Cherney AH. Prenatal Diagnosis. N. England
J. Med 1993; 328: 114-120.
7. Hoffbrand, Lewis. Tuddenham. Postgradua-te Haematology.
Fourth Edition; 1999.
8. David HK, Waye JS. Hydrops Fetalis caused by alpha.-
Thalassemia an emerging health care problem . Blood; 1998; 91: 2213-2221.
9. Higgs DR, Vickers MA, Wilkie AOM, Pretorius IM, Jarman
AP and Weatherall DJ. A Review of the Molecular Genetics of the Human
Alpha-Globin Gene Cluster. Blood 1989; 73(5): 1081-1104.
10. Kazazian HH. The thalassemia syndromes: molecular
basis and prenatal diagnosis in 1990. Semin Hematol 1990; 27: 209-228.
|

