| Acta Bioquímica Clínica Latinoamericana |
| Colestasis Intrahepática: Mecanismos fisiopatológicos
El Comité de Redacción de Acta Bioquímica Clínica Latinoamericana ha adaptado este artículo de "Medical MAG", vol. 10, Nº 96, 1999, para su difusión a través del FABA-Informa |
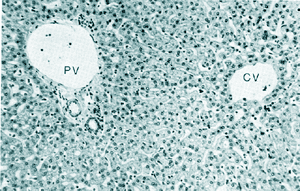
| Microfotografía con poco aumento de un corte (H y E) de hígado que muestra a la izquierda un espacio portal donde la rama de la vena porta está marcada PV, y a la derecha una vena central marcada CV. Entre los dos hay cordones de células hepáticas con sinusoides interpuestos |
| Cuadro 1 Cuadro 2 La colestasis es un síndrome clínico que se caracteriza por la disminución de la secreción biliar. Se produce colestasis intrahepática cuando la reducción se origina en los hepatocitos o en las vías biliares intrahepáticas. Este síndrome se caracteriza por la ictericia, el prurito generalizado y la elevación de los niveles séricos de bilirrubina, ácidos biliares y fosfatasa alcalina. Las causas de este síndrome son muchas, pudiendo ser agudas o crónicas y su importancia se debe a que algunas de ellas presentan una significativa morbimortalidad. En muchos casos se genera la necesidad de efectuar un trasplante hepático. En los últimos años se han realizado grandes avances en el conocimiento de la fisiología de la excreción biliar, así como de sus mecanismos regulatorios y de las anormalidades celulares que subyacen en muchas causas de colestasis intrahepática. A partir de estos conocimientos se podrán elaborar estrategias terapéuticas más racionales. El hepatocito y la secreción biliar La bilis, formada por una solución acuosa de distintos componentes orgánicos e inorgánicos, es la sustancia de secreción exógena del hígado. Se sintetiza en los hepatocitos y se excreta en las vías biliares intrahepáticas. En cuanto a su morfología, las vías biliares tienen un extremo cerrado ubicado cerca del sector central del lóbulo hepático a partir del cual se inician los canalículos biliares de alrededor de 1 micrón de diámetro. Las paredes de estos canalículos están formadas por la membrana celular de dos o tres hepatocitos adyacentes. Los canalículos biliares se ramifican y se comunican entre sí drenando finalmente en los ductos biliares ubicados en la periferia del lóbulo, los que a su vez se unen para dar origen a los conductos biliares. Se calcula en 10,5 m2 la superficie total de las vías biliares intrahepáticas del hombre. Se denomina ectoplasma pericanalicular a la región del hepatocito cercana a la membrana plasmática que bordea al canalículo biliar (membrana canalicular o apical). Esta zona se caracteriza por ser particularmente pobre en organelas, siendo ocupada por una extensa red de microfilamentos de actina (de 7 nm de diámetro) que se insertan en la cara interna de la membrana. Existen dos formas moleculares de actina que están en equilibrio dinámico: la ?actina G? es monomérica globular y la ?actina F? es filamentosa polimérica. También es posible encontrar microtúbulos de 24 nm de diámetro, dispersos en menor proporción. La red tubular está formada por filamentos intermedios constituidos por citoqueratinas que aparentemente servirían para brindar soporte mecánico a la estructura celular. En la superficie de la membrana canalicular hay numerosas microvellosidades que se proyectan hacia la luz. Toda esta estructura da lugar al denominado citoesqueleto del hepatocito, similar al presente en otras células epiteliales. El citoesqueleto es fundamental para una secreción biliar correcta pues se encarga de regular el transporte de vesículas a través del hepatocito. También genera el flujo biliar propulsando la bilis mediante contracciones peristálticas de la membrana canalicular. Formación de la bilis La bilis se forma a través de mecanismos osmóticos activos. Se cree que el 60% del flujo biliar depende del gradiente osmótico generado por los ácidos biliares mientras que el 40% restante no depende de ellos, derivando de solutos producidos tanto por los hepatocitos (glutation, bicarbonato) como por los colangiocitos. El paso limitante de la síntesis biliar es el transporte de solutos desde el hepatocito hasta el canalículo, a través de la membrana apical. Existen varios sistemas que guían a los solutos de la sangre para ser transportados hacia la luz canalicular a través del hepatocito. Se han descripto seis sistemas transportadores de iones y otros tantos que dirigen a los solutos orgánicos necesarios para el logro de secreción biliar. Algunos de ellos están ubicados en la membrana sinusoidal, en tanto que otros actúan sobre la membrana canalicular. La ATPasa sodio-potasio (ATPasa Na/K), los canales de potasio (canales K), la isoforma 1 de extrusión de protones de sodio (NHE1) y el transportador de sodio y bicarbonato (transportador Na-HC03) están en la membrana basolateral y son responsables de diversas acciones de movimiento de iones que forman parte de la bilis. Se forman así los gradientes fisiológicos a través de los cuales se mantienen concentraciones de sodio extracelulares mayores que las intracelulares y de potasio intracelular mayor que el extracelular. Por otra parte, los sistemas transportadores determinan potenciales de membrana de alrededor de -35 mV; se excretan ácidos para mantener el pH y el volumen celular, y facilitan el ingreso de bicarbonato al hepatocito. Sobre la misma membrana actúan el cotransportador de sodio y taurocolato (NTCP) y el polipétido transportador de aniones orgánicos (OATP) que cargan sales biliares conjugadas, aniones orgánicos y otros solutos orgánicos anfipáticos desde el torrente sanguíneo portal hacia el hepatocito. En la membrana apical se hallan canales de cloro (canal Cl) y la isoforma 2 de extrusión de cloro y bicarbonato (AE2) que permiten el ingreso de cloro y bicarbonato en la bilis y estimulan la secreción biliar independiente de las sales biliares. En el mismo sitio funcionan la glucoproteína-P de resistencia a drogas 1, la glucoproteína-P de resistencia a drogas 3, la proteína asociada de resistencia a drogas 2 o transportador multiespecífico de aniones orgánicos y la bomba canalicular exportadora de sales biliares. Estos sistemas, que dependen de la energía del ATP transfieren a la bilis cationes orgánicos, citotoxinas, xenobióticos, fosfatidilcolina, aniones orgánicos y sales biliares. De esta manera, el flujo biliar dependiente de solutos orgánicos se ve estimulado. También se ha determinado la existencia de un transportador de glutation, a quien se le atribuye la presencia de esta molécula en la secreción biliar. En la membrana apical de las células ductales adyacente a la luz de la vía biliar se ha determinado la presencia de un regulador de transmembrana de fibrosis quística; también se supone que existen sistemas responsables de la secreción biliar de aminoácidos y glucosa, entre otras sustancias. Importancia de los mecanismos de secreción biliar Muchas de las patologías hepáticas que producen colestasis se asocian con modificaciones del citoesqueleto del hepatocito y de los mecanismos fisiológicos de la secreción biliar. En los últimos años se han correlacionado distintas mutaciones genéticas con colestasis hereditarias que afectan los sistemas de transporte y formación de bilis. Cuando se producen cambios en el citoesqueleto se produce la pérdida de las microvellosidades presentes en la membrana canalicular, viéndose alterada la contractilidad. En modelos experimentales se ha visto que la administración de colchicina, clorpromazina y estrógenos produce alteración de los microtúbulos con disminución del transporte vesicular. Clínicamente se observan alteraciones similares a las encontradas en la enfermedad de Byler y en la cirrosis de la infancia de los indios norteamericanos. La primera fue descripta en una familia de los Estados Unidos, de apellido Byler, pero también se llama colestasis familiar intrahepática tipo I. Allí los parámetros hepáticos muestran colesterol normal, GT disminuida, sales biliares séricas elevadas, y bajo ácido quenodesoxicólico biliar. La segunda se halló en niños cirróticos de Canadá. Ambas son de muy mal pronóstico, caracterizadas histológicamente por la condensación de microfilamentos pericanaliculares. Se relacionó su aparición con una mutación en el brazo largo del cromosoma 18 que afecta la ATP-asa tipo -P. A través de diversos modelos experimentales y cuadros clínicos se ha demostrado la modificación de los mecanismos de transporte celular. Se ha podido determinar que la colestasis familiar intrahepática tipo 2 se debe a la mutación de un gen que codifica a la glucoproteína SPGP. La clínica de estos individuos es semejante a la de los que padecen la enfermedad de Byler. La colestasis familiar intrahepática tipo 3, a diferencia de las anteriores, presenta valores elevados de GT, excreción normal de sales biliares y ausencia de fosfolípidos en la bilis. Histológicamente presenta proliferación de conductos biliares e infiltrado inflamatorio en la región portal. Otra patología de la cual se ha descubierto su etiología genética asociada a mecanismos de secreción biliar es el síndrome de Dubin-Johnson. Sus parámetros bioquímicos característicos son la hiperbilirrubinemia y la excreción anormal de conjugados exógenos y endógenos. También se observaron mutaciones genéticas en la colestasis intrahepática recurrente del adulto, a pesar que se presenta de distintas manera. Todos estos hallazgos introducen la perspectiva de incluir a la terapia génica en el arsenal terapéutico de las colestasis familiares. En células ductales aisladas ?in vitro? provenientes de dos pacientes con fibrosis quística se logró suplir temporariamente la falta de CFTR usando un adenovirus como vector del gen de síntesis de esta proteína. Con la aplicación del mismo vector en ratas se consiguió que CFTR humano fuera producido transitoriamente por sus conductos biliares. Se postula la posibilidad de introducir vectores de CFTR a través de colangiografía retrógrada para el tratamiento de pacientes afectados por fibrosis quística. |